EurOPDX Galaxy
EurOPDX Galaxy
EurOPDX Pipelines
EurOPDX Galaxy bioinformatics pipelines are designed to process raw sequencing data with standardized, well known workflows allowing to export the data into formats that can be easily processed within EurOPDX research infrastructure.
Instead of filling a .xlsx template with e.g. mutation and CNA values, a PDX data provider can upload raw experimental data files acquired according to standardized procedures (currently RNAseq and Mutation are supported), together with the required data on samples, patient, diagnosis, treatment etc.
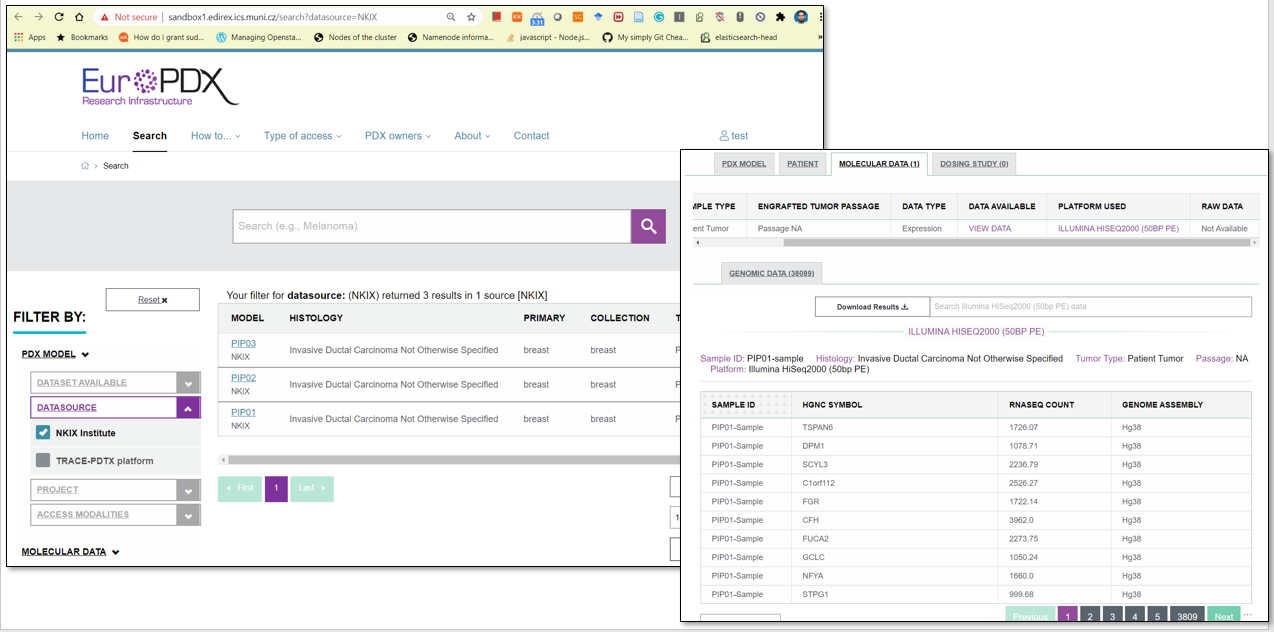
The computational pipeline processes the raw data files, deriving the quantities of interest (gene mutation, CNA values, gene expression etc.). The final step of the workflow is the upload of the results to the sandbox DataPortal, Data Hub and cBioPortal where the PDX data provider can analyze, visualize and test the data as a whole before making the resulting data set available to the other users.
EurOPDX Galaxy Pipelines are available at pipeline-prod.edirex.ics.muni.cz
Overview of bioinformatic pipeline integration

Start exploring the EurOPDX Pipelines potential now
Are you interrested in using EurOPDX Pipelines for PDX data preparation or want additional information?
Please contact us on it@europdx.eu
RNA-seq Pipeline
RNA-seq Pipeline is available in Galaxy and is integrated with sandbox EuroPDX Data Portal and cBioPortal.
Technical prerequisities
Following prerequisities are necessary to run RNA-seq Pipeline in Galaxy platform:
- Galaxy is already setup and pipeline tools with dependencies are installed
- A user account is registered in Galaxy to access it
- Reference files/side inputs are taken from ensembl (GRCh38.p13/GRCm38.p6) for RNA-Seq Pipeline - see https://www.ensembl.org/Homo_sapiens/Info/Index
- Reference files/side inputs are taken from following sources for CTP Pipeline
- https://console.cloud.google.com/storage/browser/genomics-public-data/resources/broad/hg38/v0?pli=1
- https://emea.support.illumina.com/downloads/nextera-rapid-capture-exome-v1-2-product-files.html
- Input data is Paired reads (fastq) (Note: Single-end reads will be supported as well in the near future)
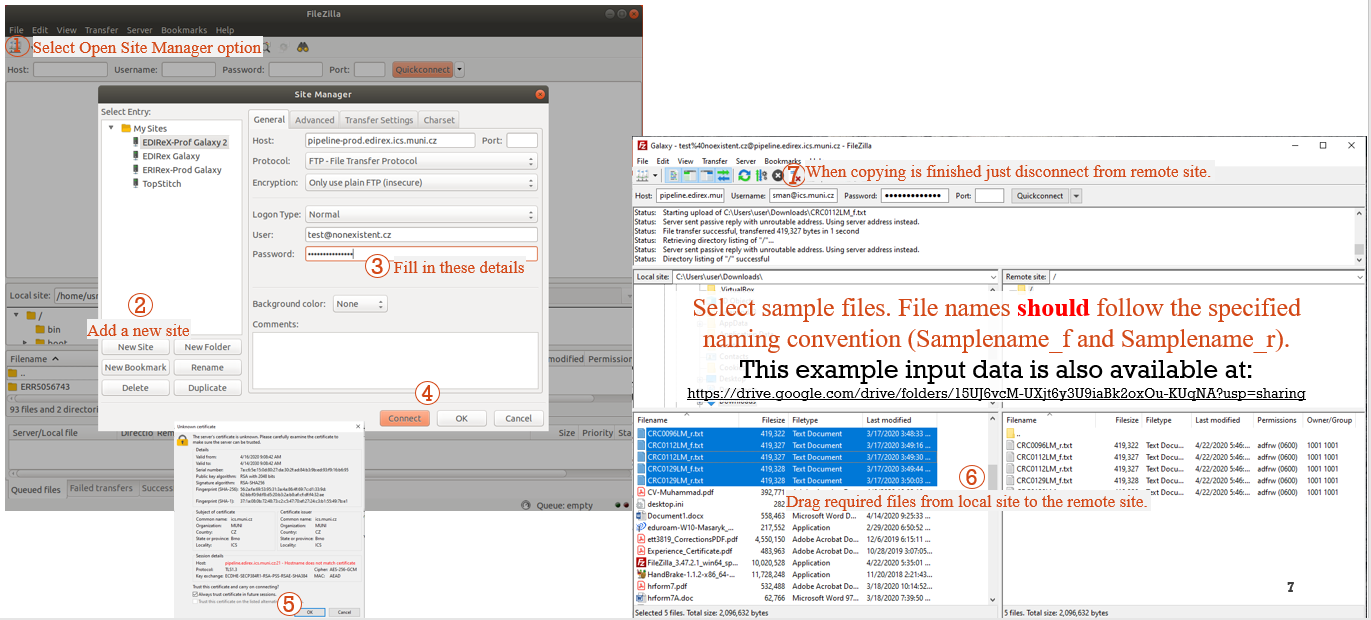
- Input data files must be named as
- For forward reads: Sample_f (e.g., PIP01_f)
- For reverse reads: Sample_r (e.g., PIP01_r)
- Example input data is available at: https://drive.google.com/drive/folders/15UJ6vcM-UXjt6y3U9iaBk2oxOu-KUqNA?usp=sharing
GALAXY CLUSTER (Openstack VM’s)

HOW TO ACCESS GALAXY
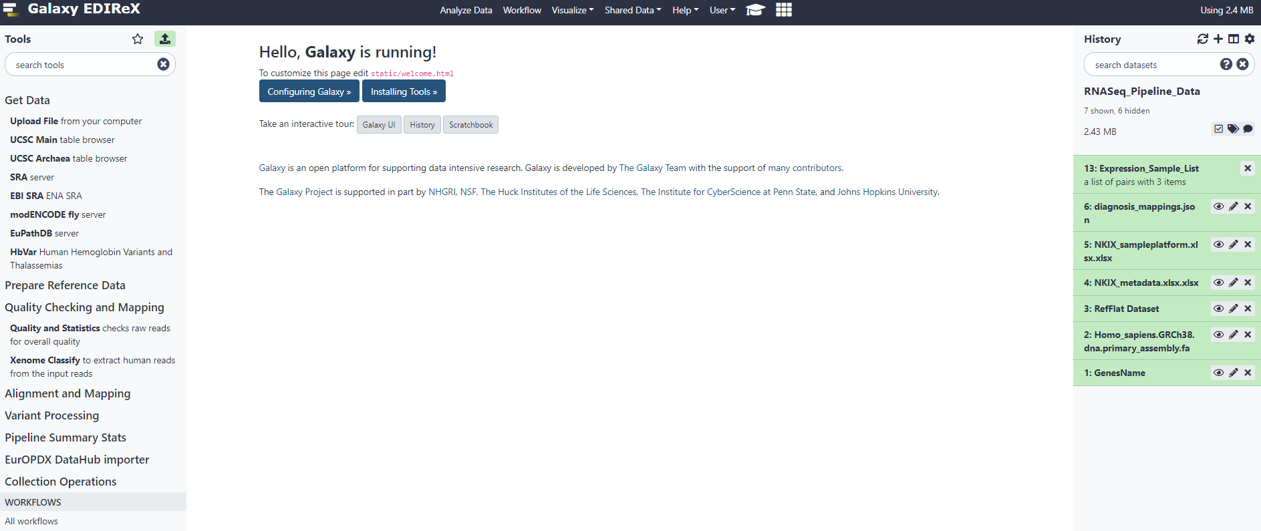
PART I: Uploading Sample Data + Metadata to Galaxy
STEP 1: UPLOAD DATA FILES TO THE GALAXY FTP SERVER
Use FTP Client (e.g., FileZilla) to upload data files to Galaxy server with following credentials:
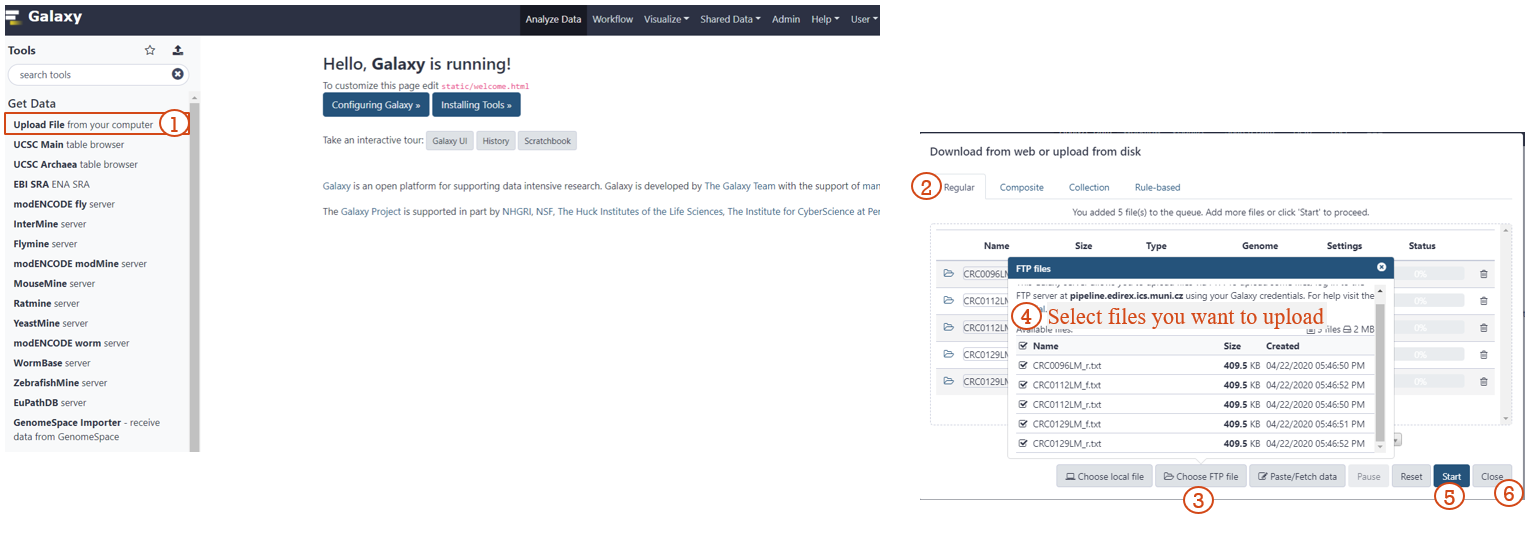
STEP 2: UPLOAD DATA FILES FROM FTP SERVER TO THE GALAXY (2/2)
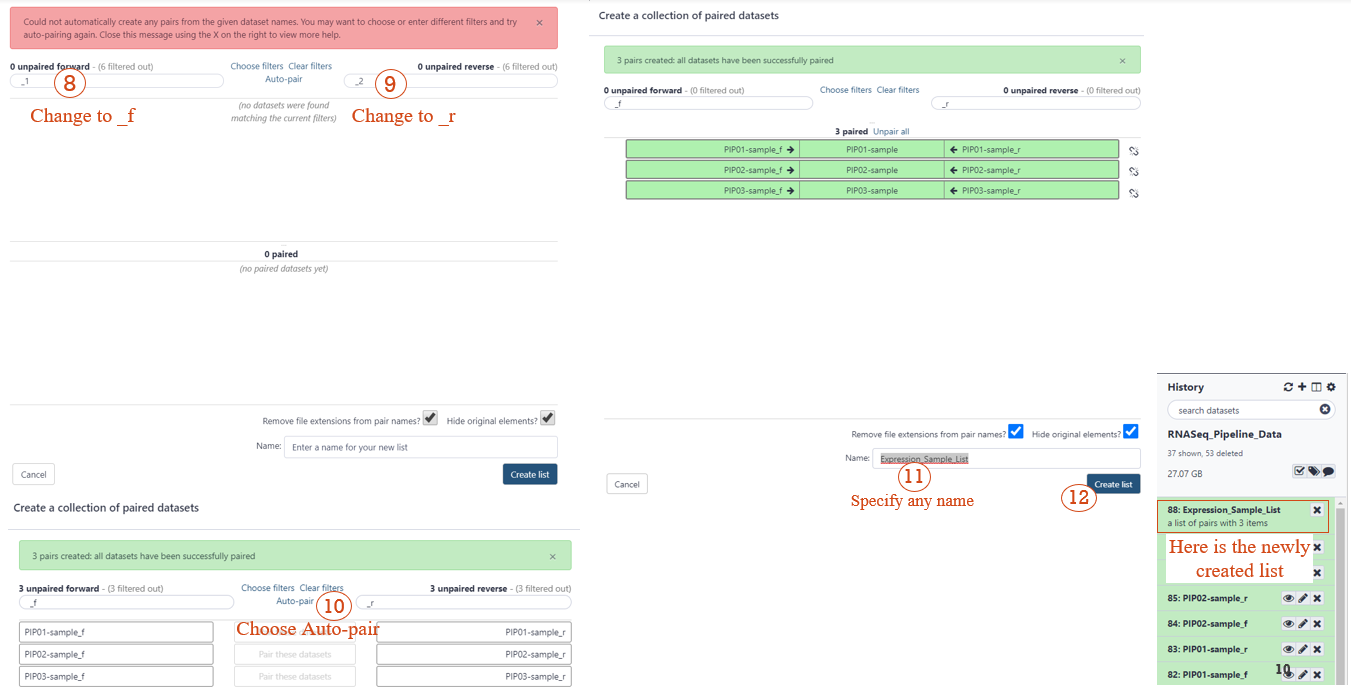
STEP 3: CREATE A LIST OF DATASET PAIRS (1/2)
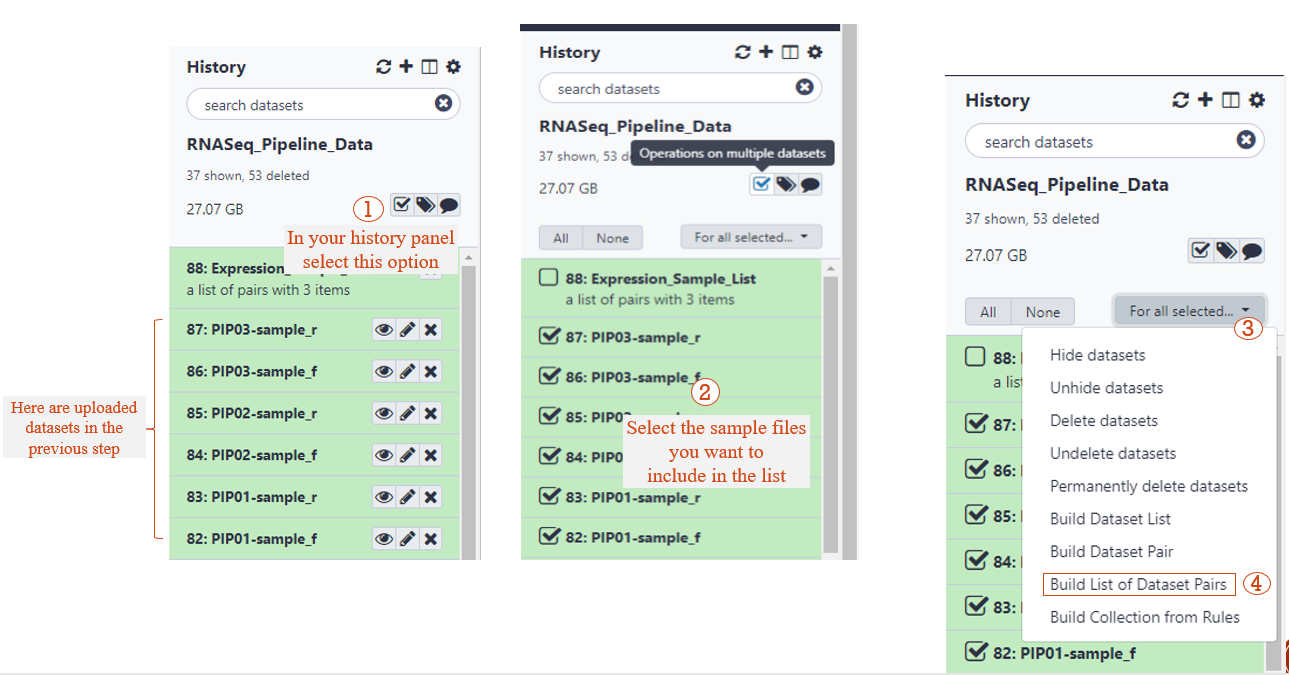
STEP 3: CREATE A LIST OF DATASET PAIRS (2/2)
STEP 4: Upload Metadata Files
- You also need following files in the correct format to successfully upload the result of pipeline execution to DataHub/DataPortal for further exploration
-
Metadata.xslx

- Sampleplatform.xslx
- Diagnosis_mappings.json
If not sure about these files than contact us for the help 😀
PART II Workflow Selection & Execution
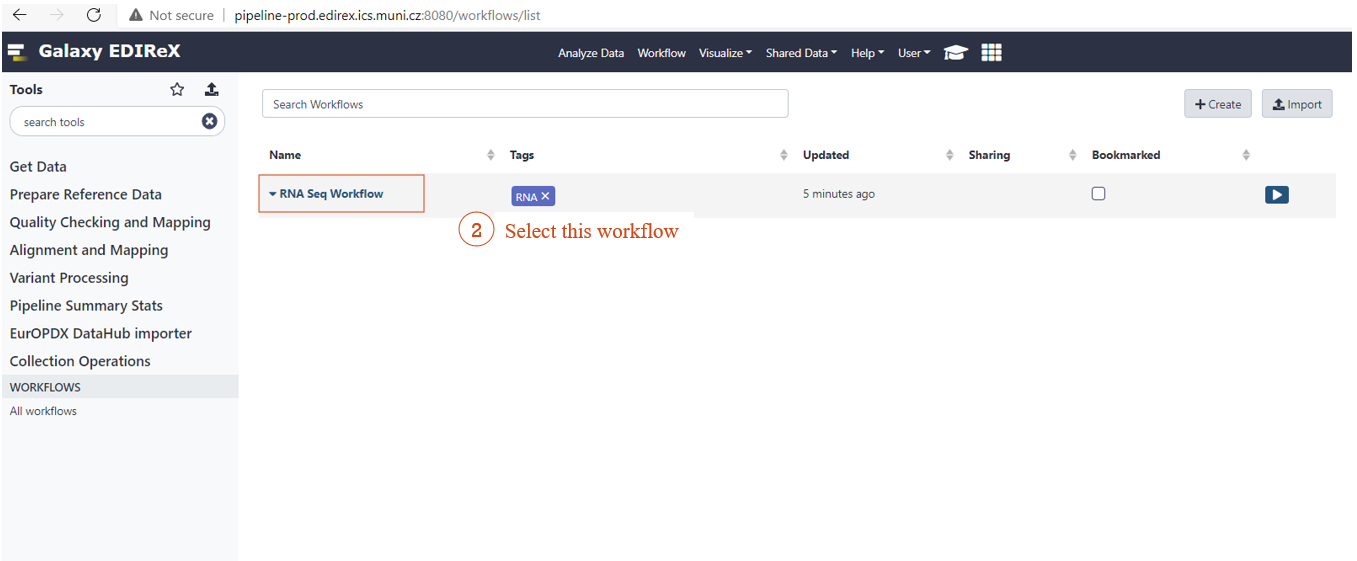
WORKFLOW SELECTION
![]() Go to this URL: http://pipeline-prod.edirex.ics.muni.cz:/workflows/list
Go to this URL: http://pipeline-prod.edirex.ics.muni.cz:/workflows/list
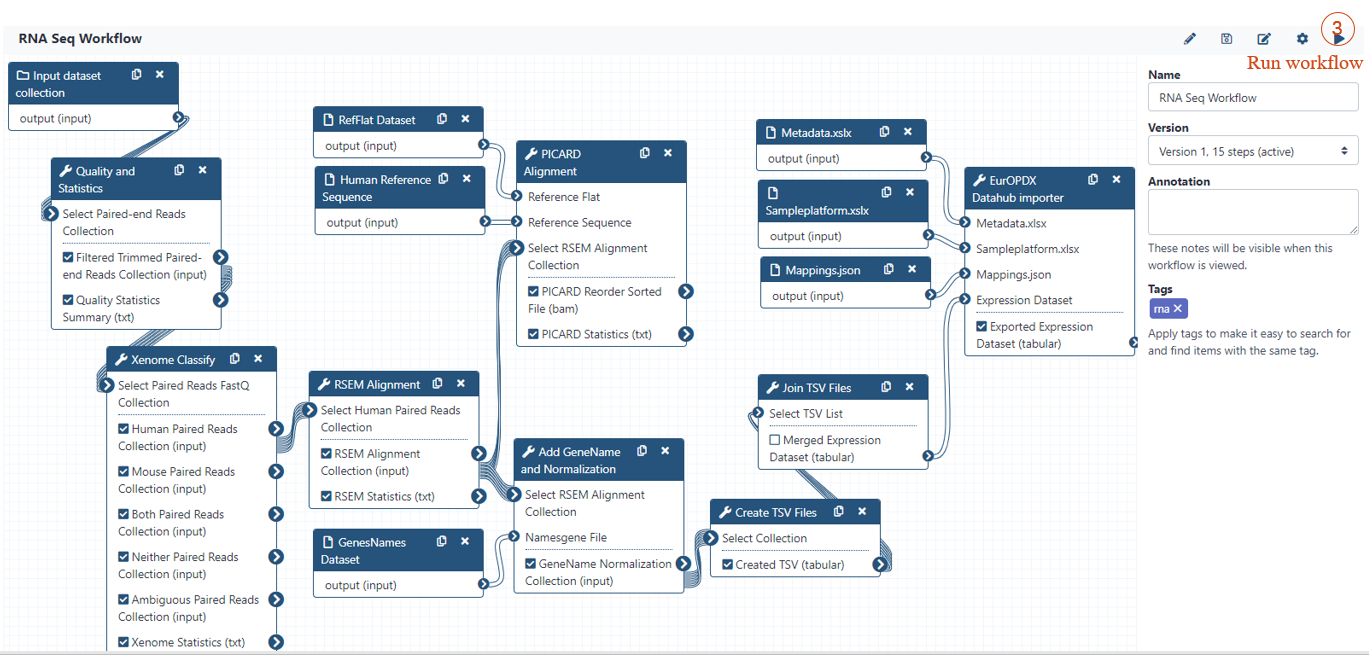
RNA SEQ WORKFLOW
INPUT PARAMETERS (1/2)

INPUT PARAMETERS (2/2)

WORKFLOW IN EXECUTION...
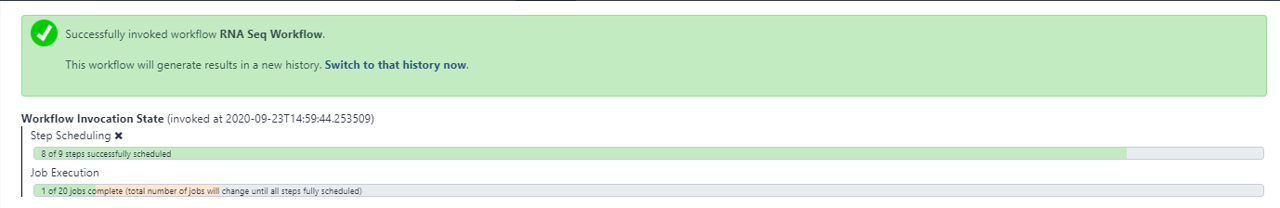
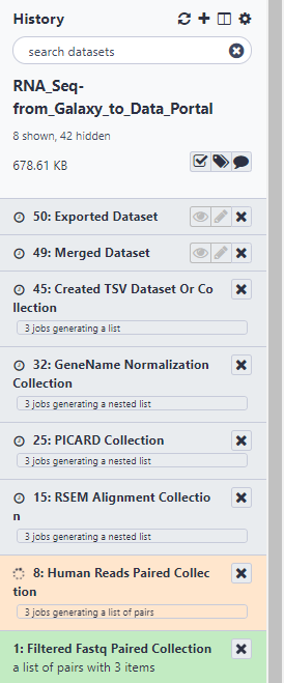
RESULTS OF WORKFLOW EXECUTION
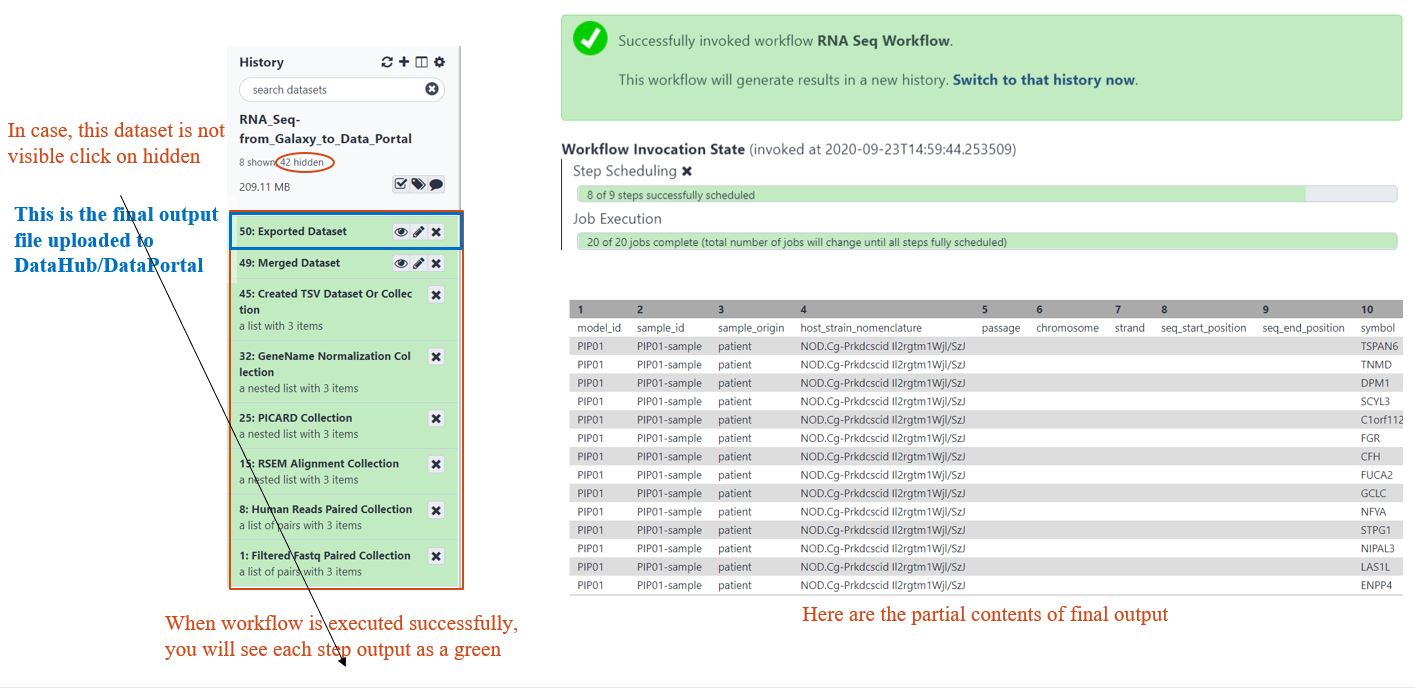
EXPLORING IMPORTED DATA IN DATA PORTAL